
We’re just one month away from the release 76 of Ensembl, which will include the long-awaited GRCh38 human assembly, and I’d like to sum up the work the comparative genomics team (aka Compara) team has been up to over the last 6 months.
Every release, we update the Compara database to include the latest genomes and assemblies. But as human is the most popular species in Ensembl, we endeavour to provide comparisons with every other species. It means that a very large proportion of the database needs to be updated due to the new human assembly.
We were able to start recomputing the alignments in February, once the assembly was loaded and repeat-masked by the genebuilders. For the homologies, we had to wait for all the gene sets (not only human, but also amazon molly and olive baboon), which happened at the beginning of the month.
Why does it take that much time to update a single species ?
So, first of all, we had to recompute more than 65 pairwise alignments. Given that an alignment takes 1-2 working days, you can imagine how long and tedious the work was. It overall represents 12.5 CPU-years of computation that was done on the Wellcome Trust Sanger Institute compute farm.
We also have a number of multiple alignments that include human (i.e. primate EPO, eutherian mammal EPO, eutherian mammal EPO-2X, amniote Mercator-Pecan). They all had to be recomputed in light of the GRCh38 assembly, but also due to the other new primate genome (the olive baboon). That’s 3.5 more CPU-years.
On the other side of Compara, the homology resources are bringing together all the Ensembl gene sets. Should a single species get updated, we have to recompute all the gene trees, orthologues, and families. That’s 20.5 CPU-years of computation, mostly spent on blastp searches.
If you add up the remaining analysis, scripts, smaller pipelines that we had to run this release, this makes about 38 CPU-years to update the Compara resources to GRCh38. I think it’s a sign 😉
How did we do that ?
All of those pipelines are beautifully managed by the eHive production system, which allow us to control and analyse complex workflows.
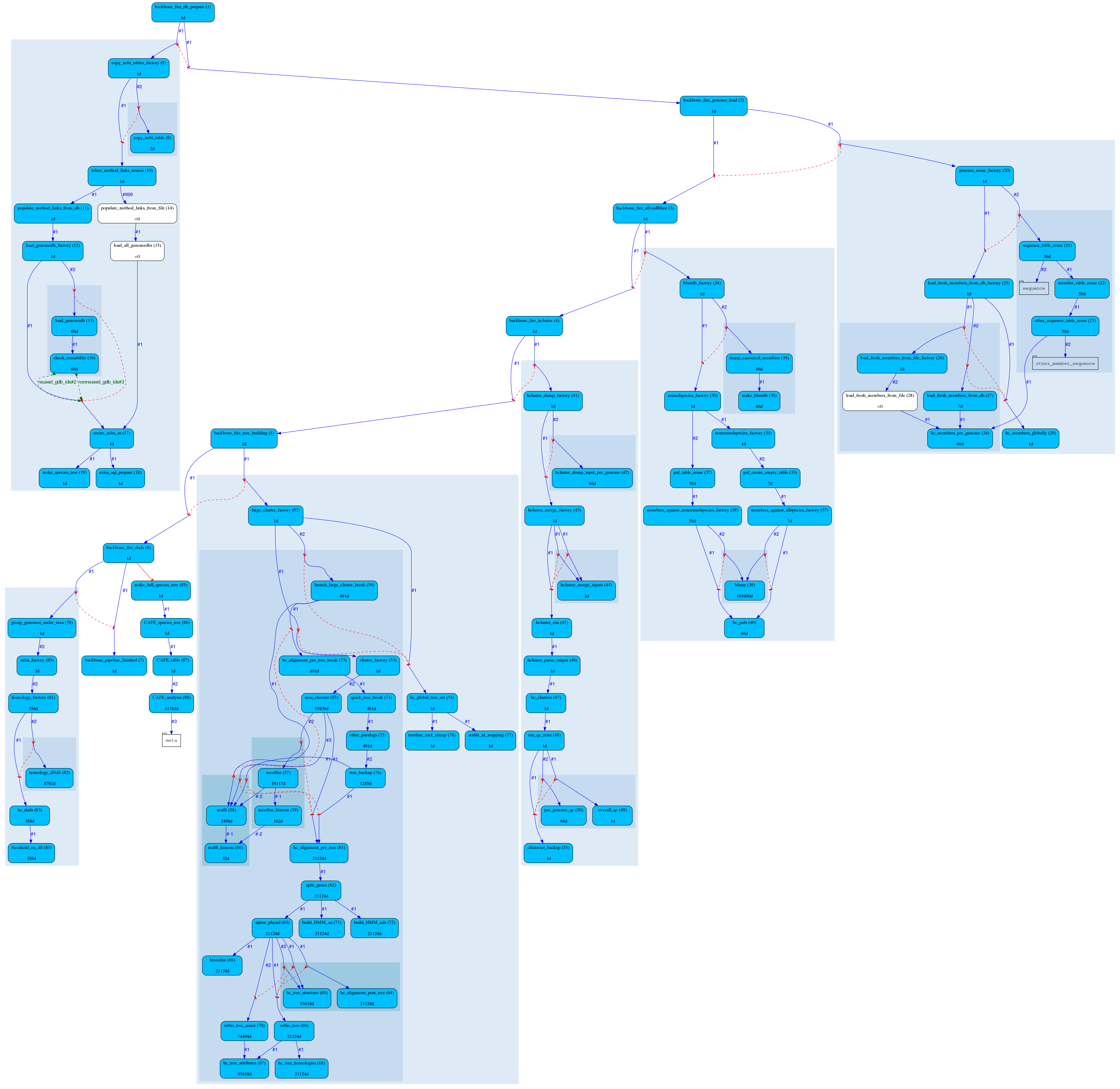

Most of the time, we only have to configure our pipelines with the names of the species, and eHive will run and manage them. Take a look at our workshop materials if you’d like to know more about it !
Great, I want the data ! Where is it ?
We’ve just released dumps of the pairwise alignments in MAF format on our FTP site. Due to the high number of I/O it required, it actually took more time to generate the dumps than to compute the actual alignments !
We are building up the Ensembl Compara database as I’m writing this post, and it will be released as part of the main Ensembl 76 release, due at the end of July. That includes MySQL dumps and programmatic access through our Perl API, as long as FTP dumps in various standard formats: BED (constrained elements), EMF (whole genome alignments, and gene multiple alignments), OrthoXML (homologies) and PhyloXML (homologies and gene multiple alignments).