A mysteriously common debilitating genetic disorder. A deadly tropical disease. One of my favourite stories in the history of genetics weaves together these two elements – it’s a good one and it always deserves a re-telling – that of malaria and sickle cell anaemia.
This story captures my attention and inspires me in the power of scientific observation, curiosity and experiment. I’m sure you are all aware of the details of this worn-out tale: it is used as an example in classrooms and lecture theatres every year to explain Mendelian genetics, haploinsufficiency, physiology, disease and protein structure and function to young scientists. To mark the coincidental coinciding of DNA day and Malaria day, we wanted to re-visit this ‘historical’ example of how scientific observation and experimental approaches have led to the understanding of how a disease as debilitating as sickle cell anaemia paradoxically persists in the human population.
Molecular biology and bioinformatics have transformed the face of biological research over the last few decades. The speed that scientists can sequence and analyse DNA means that global collaborations that study thousands of individuals are beginning to shed light on a range of different diseases.
Sickle-cell anaemia is a disease in which red blood cells form an abnormal crescent (or sickle) shape. It is an inherited disorder, and was the first ever to be attributed to a specific genetic variant (rs334, see it here in Ensembl).

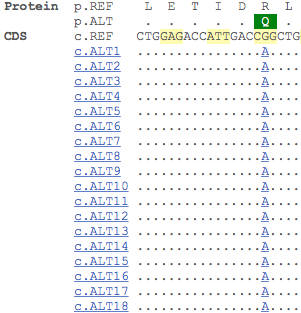
In 1949, ‘Sickle Cell Anaemia, a Molecular Disease’, from Pauling et al. identified a difference in the electrophoretic mobility between haemoglobin from healthy individuals and those with sickle-cell anaemia caused by a change in molecular structure of haemoglobin responsible for the sickling process [1]. The genetic variant (A, Reference:T) that causes cell sickling results in the substitution of a conserved glutamic acid residue at position 7 in beta chain of haemoglobin to a valine [2].
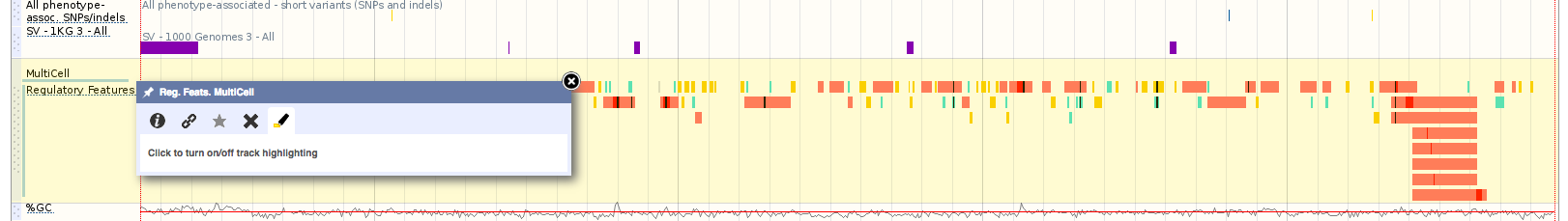
You can find this information in the Genes and regulation section for this variant. In the table below, which has been filtered to see only missense variants, the ‘Allele (transcript allele)’ column describes the variant allele (A) and the transcript allele (T, as the HBB gene is located on the reverse strand). You can also see the nature and location of the variant on the transcript in the ‘Position’, ‘Amino acid’ and ‘Codons’ columns. The SIFT and Polyphen algorithms predict the effect of the amino acid change on protein structure and function. Interestingly, only the SIFT algorithm predicts that the T/A variant would have deleterious effect on haemoglobin structure and function, confirming that predictions can never be as accurate as experimental evidence.

Only those individuals that are homozygous for the variant allele develop sickle cell anaemia, although heterozygous individuals do have the much more manageable sickle cell trait. If untreated, individuals with sickle cell anaemia have a shorter than normal life expectancy, experiencing lethargy and breathlessness throughout their lives, with increased risk of stroke and pulmonary hypertension, as well as increased vulnerability to infection. Individuals with the milder sickle cell trait can experience problems in low oxygen or as a result of severe physical exercise, but can mostly be expected to live normal lives.
As such it would be expected that this variant would be rare in human populations. However, observations made in mid-20th century revealed that this variant is, in fact, surprisingly common in African, African American and Caribbean populations (you can see this in the 1000 Genomes allele frequencies available under Population genetics in Ensembl). Coincidentally, these were people descended from those who came from areas where malaria is prevalent [3]. Why was this happening?

Individuals carrying just one copy of the variant allele were known not to develop sickle cell anaemia, leading rather normal lives. However, it was found that these same individuals, were in fact highly protected against malaria. It turned out that, quite bizarrely, having alternate alleles at this loci simultaneously prevented infection from the malaria parasite with entirely manageable sickle manifestations! Therefore, individuals with one copy of each allele have a greater chance of survival in geographical areas where malaria is endemic, preserving both alleles in the population.
Understanding this relationship has led to a deeper understanding of the infective lifecycle of the malaria parasite and novel approaches in combating malaria [4-5], but also an appreciation of the genetic factors leading to sickle-cell anaemia.
This story exemplifies how observation, epidemiology and scientific investigation can uncover the mysteries of a human disease and provide important insights for its treatment. Nowadays, this gold standard of studying single genetic disorders has been multiplied and sped up on an unprecedented scale. There are now numerous projects that are aimed at sequencing the DNA of many individuals with different diseases and using the power of bioinformatics to analyse how genetic variation might lay at the foundations for previously poorly understood diseases.
[1] Pauling L. et al. Sickle cell anemia a molecular disease Science, 1949 Nov 25;110(2865):543-8
[2] Ingram VM et al. Abnormal human haemoglobins. III. The chemical difference between normal and sickle cell haemoglobins Biochim Biophys Acta 1959 36: 543–548
[3] Allison AC et al. Protection Afforded by Sickle-cell Trait Against Subtertian Malarial Infection 1954 Br Med J 1 (4857): 290–294
[4] Mounkaila A. et al. Sickle Cell Trait Protects Against Plasmodium falciparum Infection American Journal of Epidemiology, 2012 176 175-185
[5] Gregory LaMonte et al. Translocation of Sickle Cell Erythrocyte MicroRNAs into Plasmodium falciparum Inhibits Parasite Translation and Contributes to Malaria Resistance Cell Host & Microbe, 2012 12 187-199